自己看的遗传学(四):基因组和DNA序列分析
发布时间:2021-03-19 09:08|
栏目:本章综合 |
浏览次数:
因组就是一个细胞或病毒所携带的全部遗传信息或整套基因,包括每条染色体和所有亚细胞器的DNA序列信息。研究基因组学科的就是基因组学。本篇主要分成两部分:基因组构成和基因组学研究方法(包括测序)。虽然分节很少,但是本篇的内容很长,找到加黑体句子附近的文字大略地浏览一下,可以说是很有效率的方法。
1.基因组构成
生物体基因组由整套染色体组成,一条染色体中有一个双链DNA分子,DNA分子里面的核苷酸排列顺序分别构成了基因和基因外的结构单元。生物体的单倍体基因组(生殖细胞的染色体数目是体细胞的一半,故称单倍体。此处就指的是这个概念)所含DNA的总量就叫做C值。每种生物都有自己特定的C值。即使是同一类生物中,不同种的基因组大小也有很大的差别。在一些低等的真核生物中,C值一般是随着生物的进化而增加的,比如蠕虫的C值大于藻类,藻类的C值又大于细菌——因为比较复杂的生物体,需要更多的基因去控制性状。然而,爬行类和棘皮动物的C值大小几乎和哺乳动物相等,而有一些显花植物和两栖类动物的C值甚至比哺乳动物还大得多。这就是著名的C值反常现象,也叫C值悖理:C值往往与种系进化的复杂程度不一致,生物基因组大小与生物在进化上所处的地位高低无关。但是编码每类生物所需要的最低DNA含量(最小基因组)基本上和生物在进化上所处的地位相对应——进化地位越高,形态结构越复杂,最小基因组也越大。
基因组的DNA分子可以从不同的角度进行分类。第一种分类方法,可以分成基因序列和非基因序列。基因序列就是指基因组中决定蛋白质或者RNA产物的DNA序列,一端为ATG起始密码子,另一端则是终止密码子。当一个DNA序列以ATG为起始密码子开始,随后是一个个三联体密码子,但是还未发现与这个序列相对应的蛋白质产物,此时这种DNA序列在遗传学上就叫做可读框(ORF)。一般来说,一个可读框相当于一个基因,只是产物尚未被发现或者证实。非基因序列就是除了基因之外基因组所有的DNA序列,以两个基因之间的居间序列为主。值得一提的是,可读框的定义可以被放宽到“一组连续的含有三联体密码子的能够翻译成为多肽链的DNA序列,由起始密码子开始,到终止密码子结束”(朱玉贤《现代分子生物学第4版》的定义),进一步也可以被放宽到“从mRNA的起始密码子AUG开始到终止密码子为止的连续核苷酸密码所对应的基因序列”(刘祖洞《遗传学第3版》定义)。这些定义都是可以被接受的。
第二种分类方法,基因组DNA可以分为编码序列和非编码序列。编码序列是编码RNA和蛋白质的DNA序列,所有不属于编码序列的统称为非编码序列。基因是由内含子和外显子组成的,内含子就是基因内的非蛋白质编码序列。
第三种分类方法,可以分为单一序列和重复序列。基因组里只出现一次的DNA序列就叫单一序列,比如基因序列就多半是单一序列,但并非全部都是。重复序列指基因组里重复出现的DNA序列,有的散在分布,有的聚集成簇。根据重复频率,重复序列又可以分成轻度重复序列(单倍体基因组里一般只有2~10份拷贝,结构基因基本上都属于此类。2~3份拷贝有时可被视为单一序列)、中度重复序列(10~10000份拷贝,常是非编码序列,也包括各种rRNA、tRNA和某些结构蛋白基因,比如组蛋白基因)、高度重复序列(成千上万份拷贝,例如卫星DNA等非编码序列)。原核生物基本上是没有重复序列的,真核生物中重复序列则占据着相当的比例。
同一类生物中基因组大小主要差别在于重复序列,即某一个DNA序列在基因组内有不止一个拷贝。各种不同的序列的总长度叫做序列复杂性,序列复杂性的高低反映了序列所包含的遗传信息量有多少。基因组的复杂程度还取决于基因的外显子的多寡。所谓外显子,就是指编码蛋白质或者RNA的DNA序列——这个解释并不算完整,此处只是为了帮助理解,以后还会继续提到。原核生物的基因基本没有外显子和内含子之分。
重复序列家族包括基因和基因以外的序列,以非基因序列为主,多半来源于RNA介导的转座过程。在真核生物基因组中,来源相同、结构相似、功能相关的一组基因可以被归入一个基因家族。这些重复序列一般来源于RNA介导的转座过程,即来源于反转录转座子。转座是遗传信息从一个基因座转移到另一个基因座,这个过程是由可移位因子介导的遗传信息重排。根据重复序列在基因组中的组织形式,可以分成串联重复序列和散在重复序列。
简单重复序列,就是指简单序列的重复,是DNA在复制期间通过滑移而产生的完整或不完整的短串联重复序列。它在连锁分析中非常有用,可以作为标记。简单重复序列有以下几种类型:1,卫星DNA,高度重复的DNA序列,氯化铯介质中密度梯度离心可以将卫星DNA和其它DNA分开,形成含量较大的一个主峰和高度重复序列小峰,后者就是卫星DNA区带。不同物种的卫星DNA区带数量往往不相同。很多卫星DNA序列位于染色体的着丝粒部分,也有一些在染色体臂上。这类DNA的CG含量和密度都比较低,是高度浓缩的异染色质组成部分,通常属于串联重复序列,并不转录。卫星DNA可以进一步分成微卫星DNA(2-20bp)和小卫星DNA(几百bp)。2,倒位重复序列,两个序列的互补拷贝在同条DNA链上反向排列,两个互补拷贝可以通过共价键连接,在一条DNA分子内形成碱基配对的“发夹”结构。这样说不够直观,我举一个例子,A链的核苷酸顺序从5'端到3'端,是GCACTTC……GAAGTGC,B链的核苷酸顺序则是A链从3'端到5'端的倒序,即CGTGAAG……CTTCACG。这样的序列可以让单条直线形DNA链发生折返。3,多聚dT-dG家族,类似于TGTGTGTG这样嘌呤和嘧啶交替排列的双核苷酸串联重复,平均长大概40bp,在人的基因组中大量散布。这种类型的序列往往造成DNA双螺旋呈左旋,形成Z-DNA。
散在重复序列,则可以分成四种类型:1,长散在重复序列(LINEs),是分散分布在基因组中单元长度6000bp左右的重复序列,在人基因组中有上万份拷贝。LINEs中有许多亚家族(3’非编码区不同),绝大多数是L1重复序列,L1重复序列在人和小鼠的基因组中都更倾向于分布在性染色体上。2,短散在重复序列(SINEs),是分散分布在基因组中单元长度300~500bp左右的重复序列,在人基因组中只有Alu重复序列这个亚家族是有活性的。3,长末端重复序列(LTR)的反转录病毒样因子,哺乳动物的LTR因子是来源于脊椎动物专一的反转录转座子的一个分支。最初的反转录病毒可能来源于内源性反转录病毒样因子(ERV),最后ERV渐渐减少,在人类基因组中接近灭绝。今天人类基因组中的内源性反转录病毒因子来自反转录病毒的感染。4,DNA转座子,这是通过病毒或细胞内寄生物作为载体而水平转移到新的宿主基因组的一种基因序列。由于是通过病毒之类进行转座,所以这种行为可以被动物的免疫系统阻止。
本小节在此作出总结。真核生物基因组的特点有很多,其中最突出的特点就是含有大量的重复序列,如前文所述,而且编码蛋白质的功能DNA大多数被不编码蛋白质的非功能DNA所分隔开。这就是所谓的“断裂基因”,也就是指真核生物的基因有内含子结构。因此,真核生物的基因组特别庞大,存在着大量的DNA多态性,一般来说远大于原核生物的基因组,而且绝大多数(90%以上)都是非编码序列。DNA多态性,就是指DNA序列中发生变异而导致的个体间核苷酸序列的差异,包括单核苷酸多态性(SNP)和串联重复序列多态性等等。除此之外,真核生物基因组存在着大量的顺式作用元件,包括启动子、增强子、沉默子等,这些概念本篇暂且不提,留到《遗传信息的调控》篇讲述。真核生物基因组还具有端粒结构。端粒结构是一种DNA序列和蛋白质的复合体,见《染色体》篇,本篇不再详细描述。最后要提到,真核生物基因组的转录产物是单顺反子,“单顺反子”的概念将留待《遗传信息的传递》篇阐述。
原核生物基因组则很小,大多数只有一条染色体,DNA含量也很低。原核生物基因组的特点有:结构简练,基因组中DNA分子绝大部分是用来编码蛋白质的,不转录的部分通常是控制基因表达的元件,很少冗余(真核生物基因组有冗余现象);一些功能相关的RNA和蛋白质基因,会丛集在基因组特定部位,形成转录单元,可被一起转录成含多个mRNA的分子,即多顺反子mRNA;有重叠基因,即同一段DNA携带两种不同蛋白质的信息,分成三种情况——A基因完全被包含在B基因里面、A基因和B基因有部分重叠、A基因和B基因有一个碱基对的重叠。
2.基因组学研究方法
基因功能的表达,首先要转录,产生转录物。转录的定义,可以借用高中课本的说法,即“以双链DNA中的确定的一条链为模板,以A、U、C、G四种核糖核苷酸为原料,在RNA聚合酶催化下合成RNA的过程”。这是遗传信息由DNA流向RNA的过程。然后,成熟的mRNA分子中核苷酸序列被解码,并生成对应的特定氨基酸序列,这个过程叫翻译,是遗传信息最终表达实际功能的过程。所以也有很多科学家对转录物组、蛋白质组、表型组进行研究。有关基因组学研究主要分成以下几个方面:
第一个就是基因组作图,即绘制基因组的遗传连锁图、物理图、转录图和全序列图。所有的图谱都需要作图的界标或者叫遗传标记,“遗传标记”就是指可以追踪染色体、染色体的某一节段、某个基因或某一特定DNA序列在家系中传递轨迹的任何一种遗传特性。遗传标记使用得越多、越密集,得到的图谱分辨率就越高。这么讲可能有点抽象,举几个例子来说明。第一代DNA遗传标记是RFLP(限制性片段长度多态性)和RAPD(随机扩增多态DNA),前者指的是用某一种限制性内切酶切割不同个体的DNA,不同的DNA序列存在着不同的内切酶识别位点,就会产生不同长度和数目的DNA片段;后者指的是不同的DNA序列导致引物结合效率不同,这将会影响PCR扩增。
然而,这2种遗传标记提供的信息量还是不足,第二代DNA遗传标记——VNTR(可变数目串联重复片段界标)和STRP(短串联重复多态性界标)应运而生。前者指的是基因组DNA中广泛分布着单位长度6~12个核苷酸的串联重复序列,它们以头-尾或者头-头或者尾-尾的形式串联成簇。DNA某些位置上,这种重复单位数目不同。以限制性内切酶识别串联重复序列两侧,并且进行酶切,产生重复单位数目不等的片段。后者指的是长度为2~6个核苷酸的微卫星DNA重复单元,原理与VNTR相似,而且这种重复单元分布更平均、频率更高、多态性更明显,所以更适合作为遗传标记。
第三代遗传标记是SNP(单核苷酸多态性),指分散于基因组中单个碱基的差异,包括缺失、插入和替换——大多数是替换(嘌呤、嘧啶之间的置换),以CG序列上出现最为频繁,而且多半是C脱氨转换为T。绝大多数SNP位于非编码区,位于编码区的SNP叫cSNP。SNP直接以序列变异作为标记,这将大大提高基因组作图的精度。顺便提一句,SNP和点突变蛮像的,但是前者在群体中出现频率大于1%,后者小于1%,以此来作区分。以上三代遗传标记可以在除全序列图以外所有图谱中使用(全序列图的界标是每一个核苷酸)。还有2种遗传标记是构建物理图时使用的,那就是标定位置序列(STS)和表达序列标签(EST)。前者是基因组中的单一DNA序列,后者则是某一cDNA(mRNA反转录出来的DNA,没有内含子)中特有的一段DNA序列。
将多态的遗传标记作为界标,通过计算细胞减数分裂过程中,同源染色体间交叉和互换导致遗传标记重组的频率,来确定这两个标记在染色体上的相对位置,作出来的图就是遗传连锁图,简称遗传图。遗传标记之间的距离以厘摩(cM)为单位,当两个遗传标记之间的重组值为1%时,图距就是1cM。经典遗传图只能标明基因之间的相对位置,无法标明具体位置,自然也就无法直接克隆。当然,现代作图的时候,遗传图可以转换成物理图。基因在遗传图上的位置就叫基因座,每个基因座上可以有不同的等位基因。遗传图的具体做法可以参见《连锁交换与连锁分析》篇。
除了遗传图,我们还可以作物理图。物理图是以特定的DNA序列为界标(多使用STS),直接排列在基因组DNA分子上,界标之间的距离用物理长度——即核苷酸对的数量来表示,基本测量单位是Mb、kb、bp。STS只是基因组中任何单拷贝的短DNA序列,长度100~500bp。STS物理图至少需要5套整个基因组的DNA片段,各个DNA片段将作为模板,用来自不同STS界标上的序列作为引物,进行PCR扩增。STS界标足够多,那么物理图就足够精细。最精细的物理图就是全序列图,即根据全基因组DNA测序结果作的图。可以从全序列图上看到人类基因组和其它动物基因组之间的同源现象。
物理图的作图方法有两类,一类是从长到短作图,用基因组内识别序列很少的限制性内切酶作用,分离到许多大片段,再用识别序列很多的限制性内切酶作用,切成小片段排列成序;另一类是从短到长作图,一开始就通过控制酶量和反应时间等因素,用识别序列很多的限制性内切酶进行部分酶切(作用于一部分基因组DNA),产生一些互相之间有一定重叠部分的片段(即所谓“叠连群”,contig),并用PCR予以证实,这些片段可以通过两两缀连,逐渐连成长片段。第一种方法不易丢失片段,图谱较完整,但耗费人力、物力和时间比较严重;第二种方法比较精细,分辨力高,只是容易丢失短片段,造成图谱上的空档。现在我们比较常用的是第二种方法。
转录图,也叫cDNA图,也被称为表达序列标签(EST)图。实验中,可以通过得到的某一段cDNA或一个EST,筛选出全长的转录物,并且将该转录物对应的基因准确地定位在基因组上。
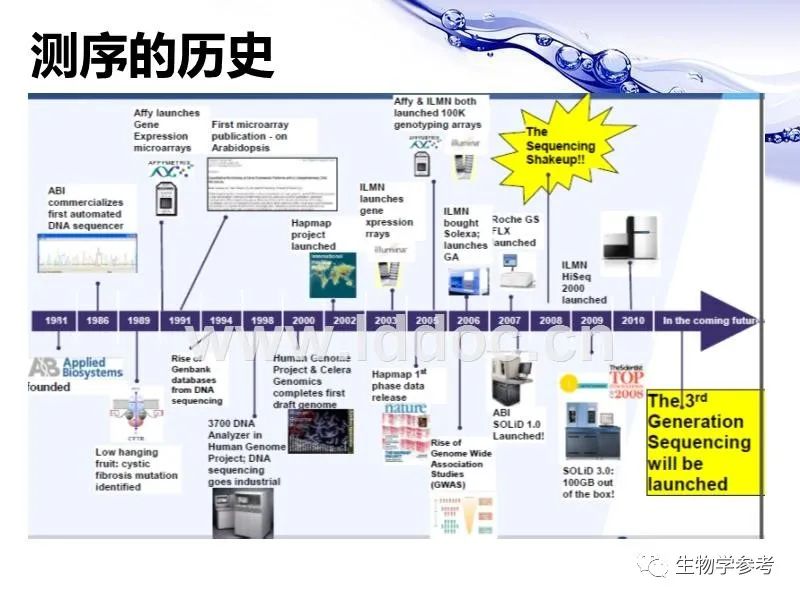
第二个是测序,即测定全基因组DNA分子的核苷酸排列顺序。实际上这也是一种作图,就是制作全序列图。测序分成手工测序和自动测序。手工测序主要是桑格法(链终止法或双脱氧法):以单链DNA为模板,用一段很短的寡核苷酸DNA当引物,加入DNA聚合酶(去掉了5'-3'外切核酸酶活性的DNA聚合酶Ⅰ的克莱诺片段),,再加入大量的dATP、dTTP、dCTP、dGTP和适量的一种ddNTP(2',3'-双脱氧核苷三磷酸,掺入到新合成寡核苷酸链3'-端会导致DNA链延伸终止),与单链模板复性,互补结合成双链。以ddATP为例,加入大量带放射性同位素磷标记的脱氧核苷酸和适量的ddATP,适当温育之后,双脱氧核苷酸不可能掺入可与其互补的每一个位置(因为脱氧核苷酸的量比较大),所以会产生出不同长度的DNA片段混合物,它们有相同的5'-末端,在3'-末端的ddATP处终止。将这种混合物加在变性凝胶上,电泳分离,可以得到一系列全部以3'-末端ddATP为终止残基的DNA片段(梯状条带)。同样的操作分别加入ddCTP、ddTTP、ddGTP,点到同一变性凝胶上电泳,然后X射线底片曝光,可以直接读出DNA的核苷酸顺序。
此外还有马克夏姆-吉尔伯特法。这是一种化学测序法,将片段的一个末端利用放射性同位素磷标记,分成四份,用特定的化学反应降解部分的DNA(对于某一种脱氧核苷酸专一)。反应产物用聚丙烯酰胺凝胶电泳分离成一系列DNA片段,X射线曝光底片读序。
测序技术进一步发展,全基因组鸟枪法测序技术出现,随机挑选带基因组DNA的质粒进行测序,在计算机的帮助下进行序列拼接。我们尝试建立高度随机、插入片段大小1~2kb的基因组文库,保证克隆数;也尝试高效、大规模的末端测序,开发新的拼接软件,建立λ文库,以填补测序缺口。尽管如此,以鸟枪法测序高等真核生物基因组(存在大量重复序列)时,还是会导致拼接错误。在处理较大的真核基因组序列分析时,通常用稀有限制性内切核酸酶先将待测基因组降解为几十万个碱基对的片段,再分别进行测序,或者根据染色体上已知基因或遗传标签的位置来确定部分DNA片段的排列顺序。
最后介绍几种新的测序技术。这些测序技术都在中国常见的遗传学教材中出现,所以在此简单叙述一下。第二代测序技术均基于桑格法进行改进。第二代测序技术主要是采用了大规模的矩阵结构芯片分析技术,说白了就是用了芯片这个固相载体。基本流程如下:先将基因组DNA随即切成小片段,然后在小片段DNA分子末端连上接头,接着变性得到单链模板文库(意思就是得到了一大堆单链的小片段模板),再将单链小片段DNA固定在固体表面,之后对固定片段克隆扩增、制成polony芯片,最终利用DNA聚合酶或连接酶,对芯片上的DNA进行循环反应,读取碱基连接产生的信号,对阵列图像进行时序分析,获得DNA片段的序列,并且通过计算机形成叠连群获得全基因组序列。第一种是罗氏(Roche)的454焦磷酸测序。454系统首先将随机片段化后的基因组DNA变性成单链并稀释,让每个磁珠通过表面引物“捕获”至多一个DNA分子。然后每个磁珠在封闭的乳胶小泡中PCR扩增至数千拷贝,再次变性去除游离的DNA分子。富集磁珠并置于光纤载片的小槽内,以4种天然核苷酸为底物合成互补链。当某个新添加的核苷酸被整合到延伸的DNA链中,它释放的焦磷酸被硫酸化酶转化成ATP,荧光素酶利用这个ATP催化荧光素释放光信号,被光纤束连接的CCD检测到。454系统主要的错误出在碱基插入和缺失,尤其是测同多聚核苷酸的片段时(因为这种片段的长度检测只能通过光信号强度推断)。
第二种是Illumina的Solexa基因组分析仪。Solexa同样先随机片段化基因组DNA,然后将各个单链模板与固相基底上的正、反向PCR引物随即杂交引起互补链合成,随后经过DNA变性引起邻近引物桥连扩增。不同模板生成的扩增子集群散布在基底的不同位置,形成阵列。测序过程中,测序引物与模板末端的共同接头序列杂交,在DNA聚合酶催化下以4种3'-OH位置加了一个可切除修饰基团的核苷酸为底物合成互补链。每一轮链延伸只有一个碱基整合,因为修饰基团会阻止下一个核苷酸整合。每个核苷酸还带有一个可切除的荧光标记基团。也就是说,每一轮链延伸,都检测一遍荧光图像信号,检测完后切除修饰基团和荧光基团,进入下一轮合成。Solexa系统主要的错误出在碱基替换。由于修饰和荧光基团的切除可能会不完全,有效读长也会偏短。
第三种是ABI公司的SOLiD。SOLiD系统采用双碱基编码探针,片段化基因组DNA后用乳胶PCR进行扩增,这些步骤和454系统类似。测序引物与模板DNA杂交后,启动八核苷酸探针与模板的杂交。从第n位碱基开始,用DNA连接酶催化连接、采集荧光信号、切除探针后三位核苷酸及所标记的荧光标签三步循环。简并八核苷酸探针的第一位和第二位碱基决定探针的荧光标记颜色。10次循环,产生10个荧光信号,对应DNA序列上每5个碱基中的前两个碱基序列。10次循环后,变性恢复单链模板,选用不同引物,从n-1位碱基重新开始10个循环,以此类推。SOLiD系统的主要错误在于碱基替换,测序通量最大,读长较短。
第二代测序技术仍然使用了体外PCR扩增,这会引入突变,也会影响序列丰度等信息。所以,第三代测序技术HeliScope单分子测序法出现了。HeliScope和Solexa原理相似,但没有模板扩增这一步,也不需要阻断链延伸的修饰基团,因为它是对于单个分子进行测序。它是用高度敏感荧光检测装置进行测序的。HeliScope的主要错误在于碱基缺失,可以通过使模板链变性并去除、对新合成链二次测序来提高准确性。
除此之外,还有片段化单分子实时荧光测序的Pacific Biosciences测序系统、构建带有荧光染料的DNA聚合酶的Life/VisiGen技术,以及尚未正式投入使用的纳米孔测序策略等等,这些都是第三代测序技术了,主要特点是单分子序列分析。新的高通量测序平台的应用,全基因组相关分析(GWAS)的发展,基因组的分析手段变得越来越全面,基因组的秘密也越来越多地展现在我们面前。还有混杂基因组学,是研究混杂基因组的——比如直接提取土壤样品中所有微生物的总DNA(因为有些微生物是难以分离培养的),限制性内切酶酶切片段克隆在表达载体中,然后去转化宿主细胞,表达出大量的产物。混杂基因组学对于研究已经灭绝的古生物和生物进化方面理论很有帮助。本篇已经介绍了一些分子生物学的内容,更多相关知识可以去了解分子生物学和基因工程相关书籍。
-

扫一扫关注于我们
-
 网站首页
网站首页 必修一
必修一 必修二
必修二 选修一
选修一 选修二
选修二 选修三
选修三 高考竞赛
高考竞赛 教材汇总
教材汇总